One of the most common analysis tools in population genetics is Principal Component Analysis (PCA), so much so that a stylized version of this analysis is featured on the cover of the popular textbook "Molecular Population Genetics" by Matthew Hahn.

However, like any computational analysis, the data do not "speak for themselves" but reflect choices that researchers and data collectors have made, and sometimes even biases that they hold. However, like any analysis involving data, the data do not "speak for themselves" but reflect choices that researchers and data collectors have made, and sometimes even biases that they hold. These choices can be rendered invisible in publications, as researchers may feel pressure to represent sparse data as robust and downplay the possibility of equifinality of results. Here I examine practices in genetic data collection and curation, and the implications of these choices on computational analysis and visualization.
Many studies have requirements for volunteers to participate and contribute. For instance, some studies have required volunteers to have all 4 grandparents born within a certain distance (e.g. 50 or 100 km) of each other. Others only allow people with “local” surnames to participate. Still, others, while they may allow anyone to contribute genomic samples to a study (usually in the form of a cheek swab or blood draw), will remove any individual genomes that are observed to be an outlier from the group in computational analysis.
What are the consequences of these practices? These participant inclusion (and exclusion) strategies can result in plots that might appear to have gaps or blank spaces, such as the PCA below, built using published genomic data that was ascertained using the above criteria.
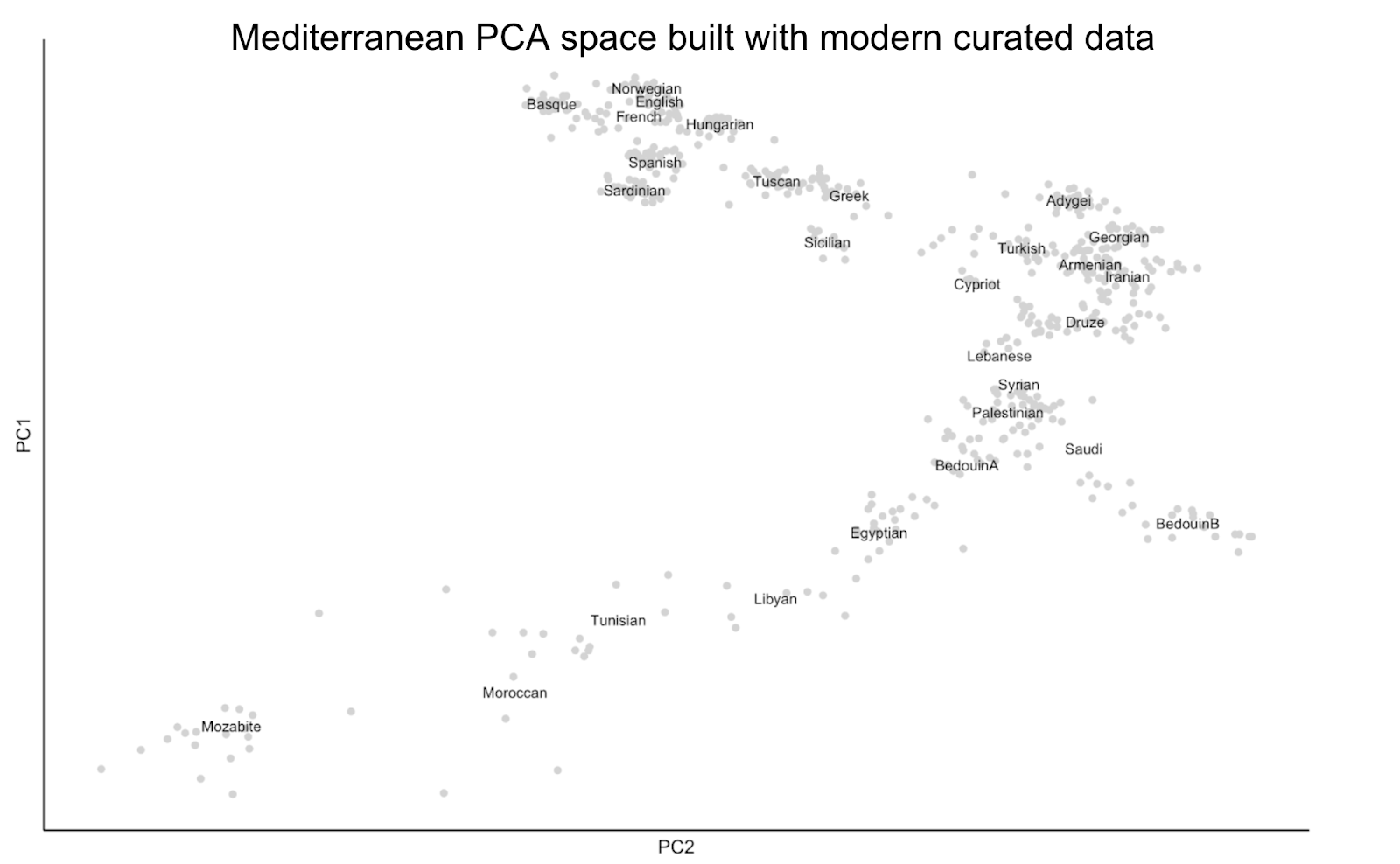
Since most genetic data is obtained and curated in this way, the impacts of such exclusions may not be easily discernible but become visible when ancient genomes are added to analyses, as they are not subject to these same inclusion criteria. In the plot below, I visualize ancient genetic data from the Etruscan site of Tarquinia, in Italy, and the North African site of Kerkouane in Tunisia, to illustrate this point. This data was published in a paper entitled “A genetic history of continuity and mobility in the Iron Age central Mediterranean” by Moots, Antonio, Sawyer et al 2023, and also includes other published ancient genomes from the Bronze and Iron Age Mediterranean.
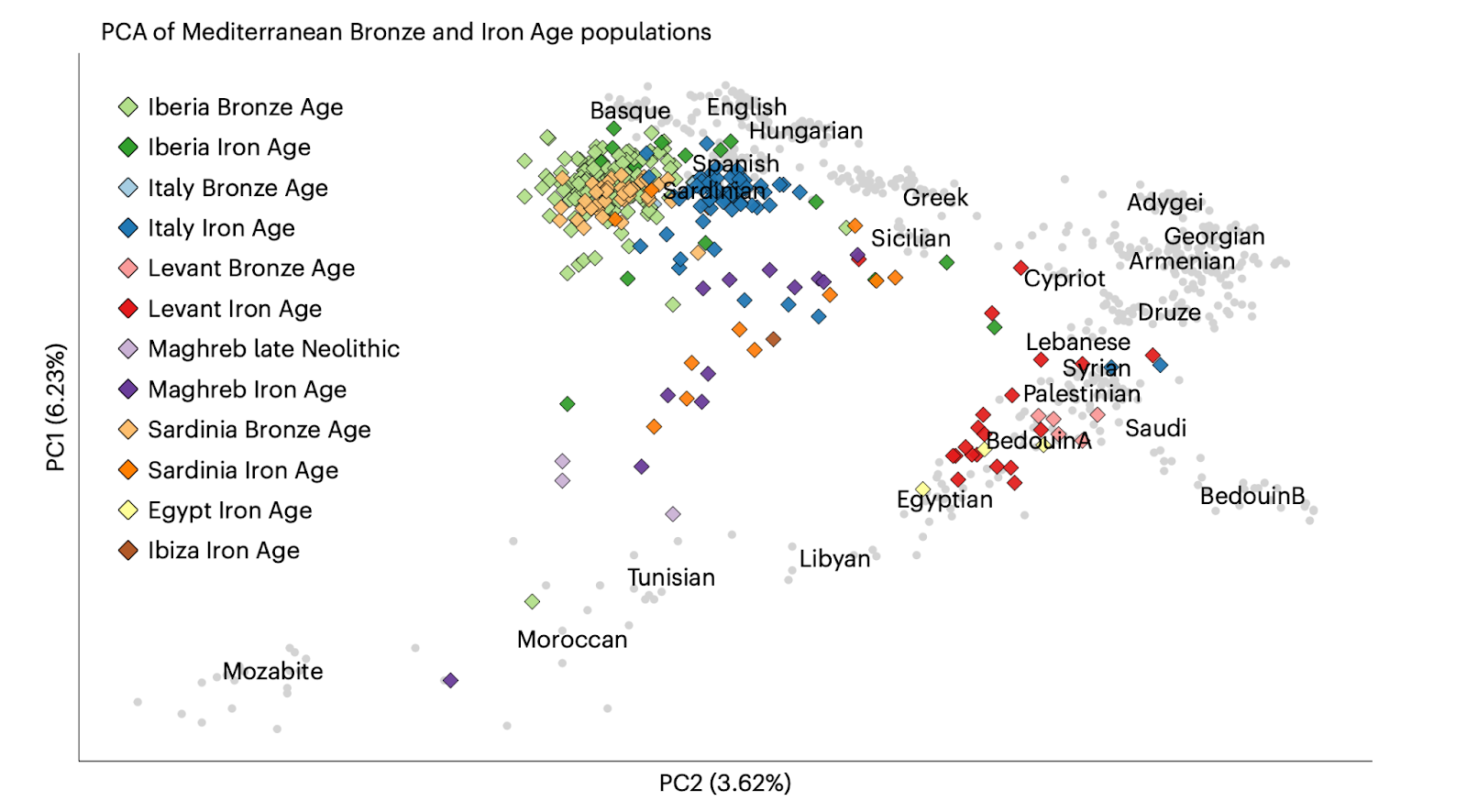
Many of these ancient genomes project into the space that was empty when only modern genomes were visualized. When inclusion in the study isn’t predicated upon grandparents’ birth location, surname, or any of the other factors used to curate modern data, we see that populations from different regions are intermixed and overlapping, rather than discrete. The field’s approach to data acquisition values data tidiness (and perhaps even purity) over representing the diversity of our communities. These ancient individuals have helped us glimpse ghosts of past approaches to population genetics, still haunting the field today.
Comments
Post a Comment